细胞生物学的相关研究一直受限于数据的完整性和表型的完整性,对应激状态和稳态下的细胞区别观察不够充分。过去五年中,计算机视觉和语音识别领域通过对大量的无标签数据进行学习、建模,很好的解决了数据不足的问题。同样在最近的研究中,机器学习方法使用单细胞数据进行扰动建模也推动了细胞生物领域前进。对于生物学家来讲,无论研究基因、转录本、修饰、蛋白功能,都要频繁的进行人为干预,实现对感兴趣变量的正向或者反向的改变,从而观察细胞表型的变化,整个过程需要对干预工具的构建、导入、实验观察,从而得出表型结论,而扰动建模的目的就是想要通过数学模型的建立,通过对已有数据的分析、归纳和总结,对一个分子的功能在没有湿实验时做出预判,对于生物学家和药物研发者来讲,好的模型一定能够帮助加深对生物机制的理解,推动药物的研发进程。
在“perturbation modeling”这个概念下,机器学习使用单细胞数据尝试处理了哪些问题?模型是如何构建的?观点来自Machine Learning for Perturbational Single-Cell Omics。

本期综述结合相关文章展开相关讨论。文章中反复出现的“perturbation”一词该如何理解?原意为干扰、扰动,具体在细胞生物学当中对应的就是细胞所受到的外部、内部干扰因素:内部干扰因素如基因突变;外部干扰因素如分子(小分子化合物、大分子等外源性分子)处理、基因knock out、knock down等干预,这些都是文中perturbation所对应的因素,因此perturbation modeling的最终目的,就是通过对已有数据的学习,准确捕获perturbation所带来的生物学意义,解释该perturbation对哪些靶点、信号通路产生影响,同时对新的perturbation类型所带来的影响进行预测,甚至当多个perturbation同时出现是协同还是拮抗做出判断,从而解决分子细胞生物学以及药物发现领域等未知的问题,perturbation一词也在后面的文章中统一翻译成“扰动”,请读者知悉。
包括基因敲除、刺激物、再生因子等对细胞处理的过程本质上都是一种对细胞状态的扰动,而系统生物学的重要分支就是对这种扰动的效果进行建模、预测。从结合(binding)到对接(docking),到下游分子效应和器官的整体改变,如果要对扰动带来的变化进行评估,必须对细胞、分子和组织之间的相关机制有全面的了解。在传统的分子细胞生物学实验中,研究者会进行大量的蛋白-药物互作、生物标志物筛查等试验方法来捕获这种外界刺激对细胞本身带来的改变。
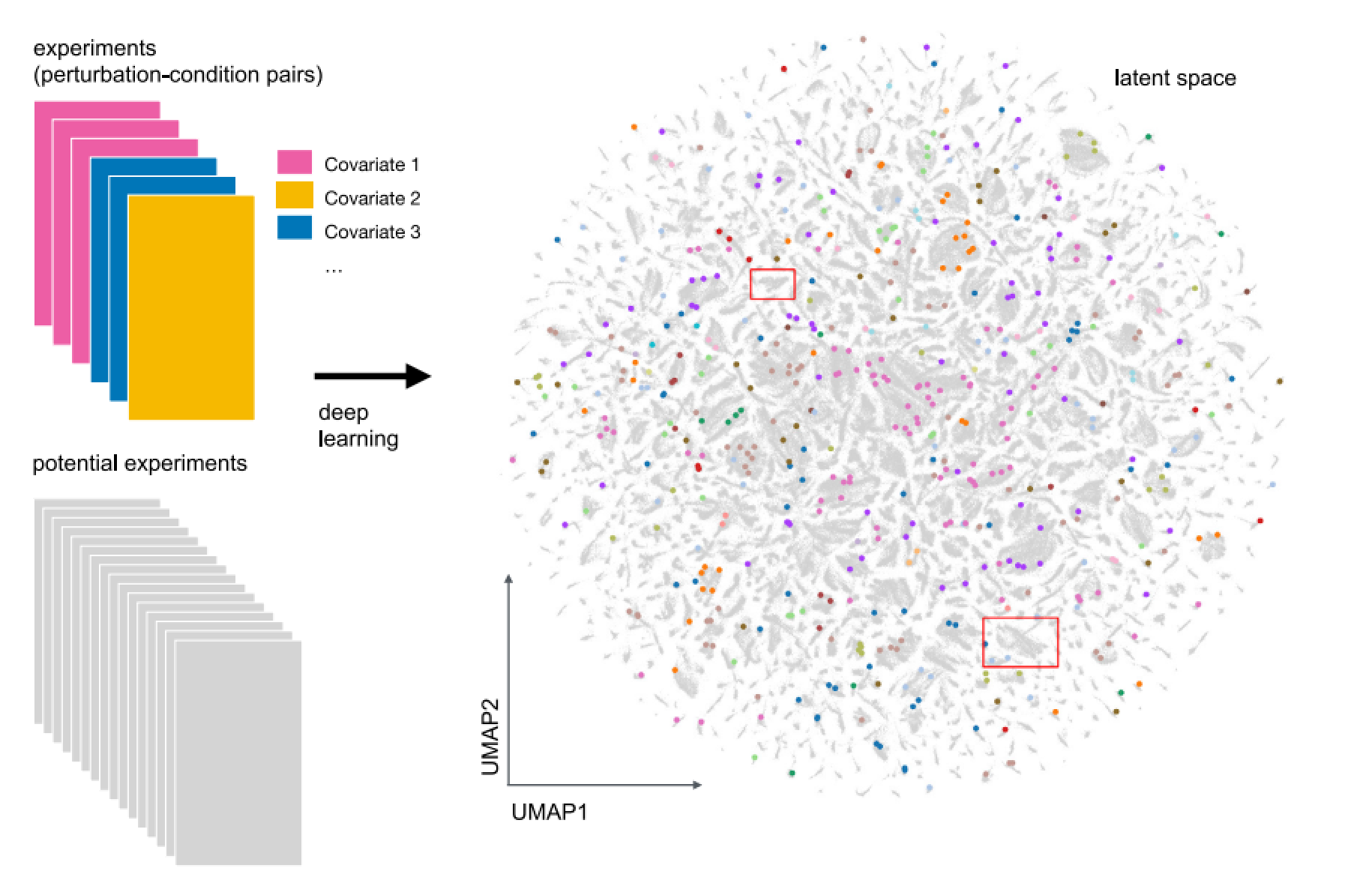
图1:扰动隐空间的建立:每个点代表一个扰动-条件组合对(扰动-条件二维矩阵中的数据点):带颜色的点代表已有实验,点的颜色代表不同实验之间的相关性,如剂量、细胞类型、通路活化等
为了批量获取这种扰动信息,很多研究单位建立了相关细胞数据库,比如体外药物筛选数据库(例如Genomics of Drug Sensitivity in Cancer、 Cancer Therapeutics Response Portal、Connectivity Map等),还有其他广谱性质的数据收集,例如PharmacoDB、DurgBank数据库针对每个细胞系或者药物建立信息体系,同时这些数据库包含了小分子化合物的一般化学特性。
除了数据上的积累,机器学习模型在过去十年中也得到了发展,不断的对越来越多的数据进行训练、开发、测试,用以获得更好的机器学习模型。经典的ML模型已经有不少用于药物靶点发现和IC50的预测。由于强大的方程拟合能力和灵活性,深度学习模型在最近的研究当中被广泛使用,通过卷积操作处理化学和序列数据,预测化合物结构毒性、通过体细胞突变预测药物敏感性等,但是通过深度学习对扰动生物学(perturbation biology)中的分子机制进行的研究并没有取得太多的成果,对于细胞扰动应答机制的理解还远远不够。
本篇文章关注最近单细胞测序的发展,讨论最近利用DL方法建立的用于处理上述问题的成功案例,单细胞测序所产生的细胞水平数据让DL模型看到了更多转录组、蛋白组和表观水平的数据变化,庞大的数据可以同时对细胞应答的异质性沿着多个variation方向进行建模,目前做的最多的,也是最前沿的研究应该是使用scRNA-seq数据进行细胞应答的建模分析。
由于单细胞数据提供了更多的训练实例和变化方向,深度学习模型可以通过更少的试验次数对一些未知、未见的事件进行推理。使用隐空间概念对分子特征进行描述就是一个很好的例证,通过这种对基因组、表观组、蛋白组特征的蒸馏表示(distilled representation),来提取、总结细胞扰动和应答之间的关系,这种空间建立过程对反复出现的事件(如信号通路多个因子的共表达、细胞生物标志物的出现)进行强调并赋予权重,从而对细胞的应答特性进行捕获。对隐空间的另外一种理解就是类似PCA、t-SNE或者UMAP一种数据压缩方式,比如,一个2维的auto-encoder隐空间表示非常类似于t-SNE,生成模型使用这个概念对细胞状态之间的互作关系进行隐式建模,使用监督学习用来压缩数据从而学习细胞状态的扰动关系。
虽然深度学习模型可以这样广泛的使用,但是这种计算模型和显微镜下的实验观察是完全不同的两件事,目前现有的大部分数据库的主要缺陷是缺少perturbation的条件。深度学习模型对数据量要求非常高,如果没有充足的数据量和perturbation实验,深度学习模型也很难解决一个这样高纬度的生物学数据的压缩问题。参考计算机视觉领域的发展历程,只要标准化数据和benchmark的出现,单细胞数据也会被源源不断的用于深度学习的模型建立当中。
扰动建模概念下,具体围绕单细胞数据尝试解决了哪些生物学问题?
1. 扰动建模的目的
扰动建模是一个大的机器学习框架,其中包含多个具体模型和任务,相应的,每个模型会有评判方法来评价该模型的表现,在这个框架下的具体任务基本上是对细胞对外界刺激应答的评估和预测。
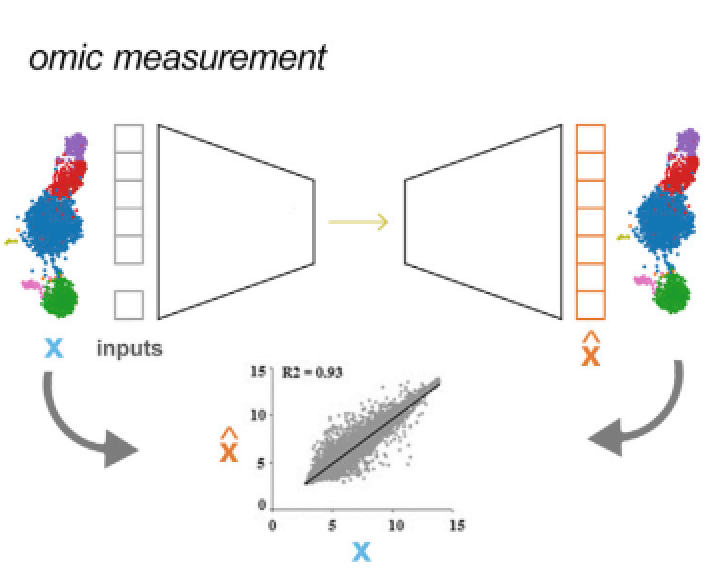
图2:组学评估: 预测转录组或者其他组学在扰动之后的变化, 模型的输入通常是分组信息(处理组、对照组)以及不同分组对应下的组学数据,模型的性能可以通过预测值 (![]() ) 同真实值 (X)之间的相关性来评估
) 同真实值 (X)之间的相关性来评估
A. 组学特征和表型预测
如果模型已经捕获了一个扰动所带来的重要生物意义,就可以在细胞受到扰动因素影响以后预测未知的组学特征和表型变化(图2),在扰动发生之后,模型可以预测转录组或其他组学的变化情况,模型的性能可以通过真实数据和预测数据的相关性来评估;半数抑制浓度(IC50)预测是表型预测的一个经典问题,模型通过学习其他细胞系IC50值的变化特征,预测目标细胞系的IC50值(图3),模型的表现可以通过经典的回归模型评价方法进行评价。通常这里提到的有关表型的预测任务就是在解决药物发现中所遇到的实际问题,比如药物有效性,针对不同肿瘤的药物敏感性的预测,表型预测其实就是在尝试解决药物发现中的部份问题。
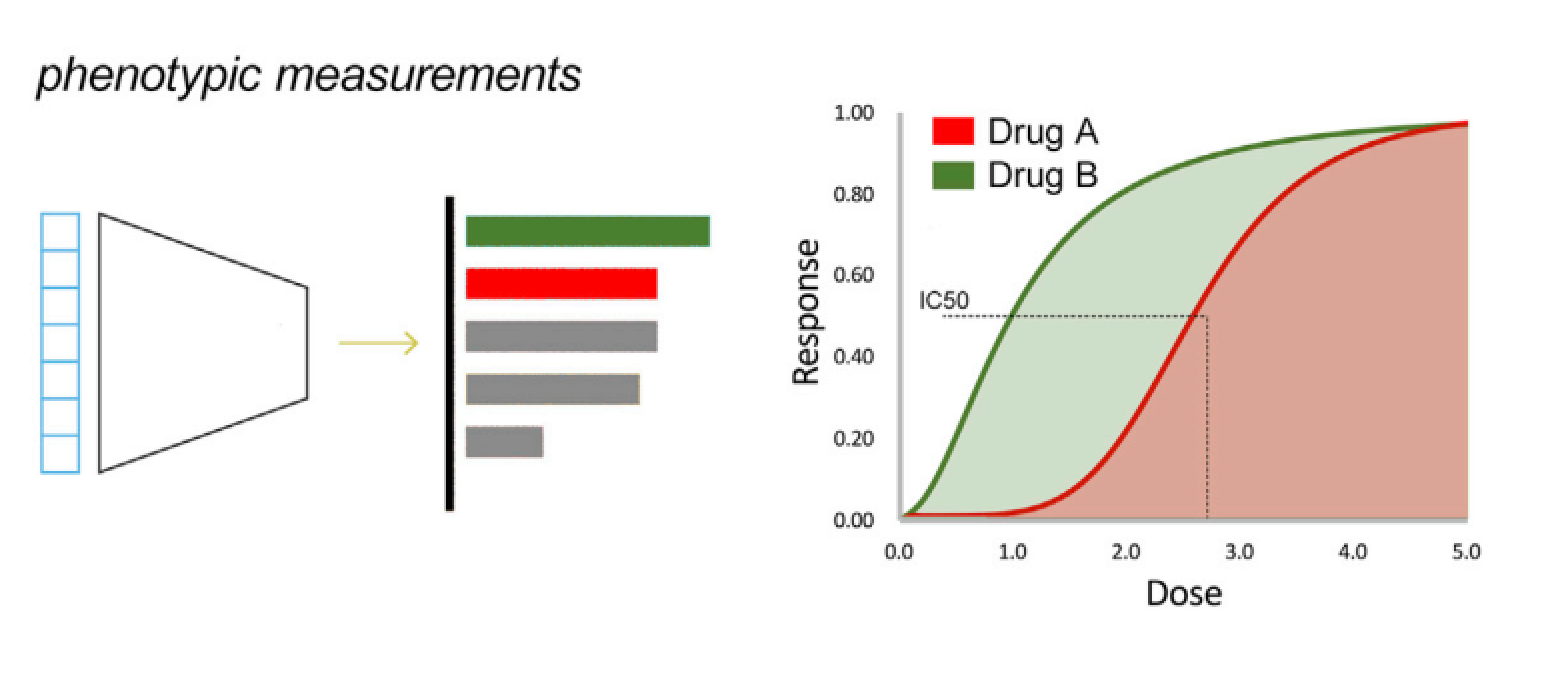
图3:表型预测: 用连续变量来估计细胞系对各种扰动的相关值变化,比如IC50,量效曲线(Dose Response Curve)、细胞毒性等指标
B. 靶点和机制预测
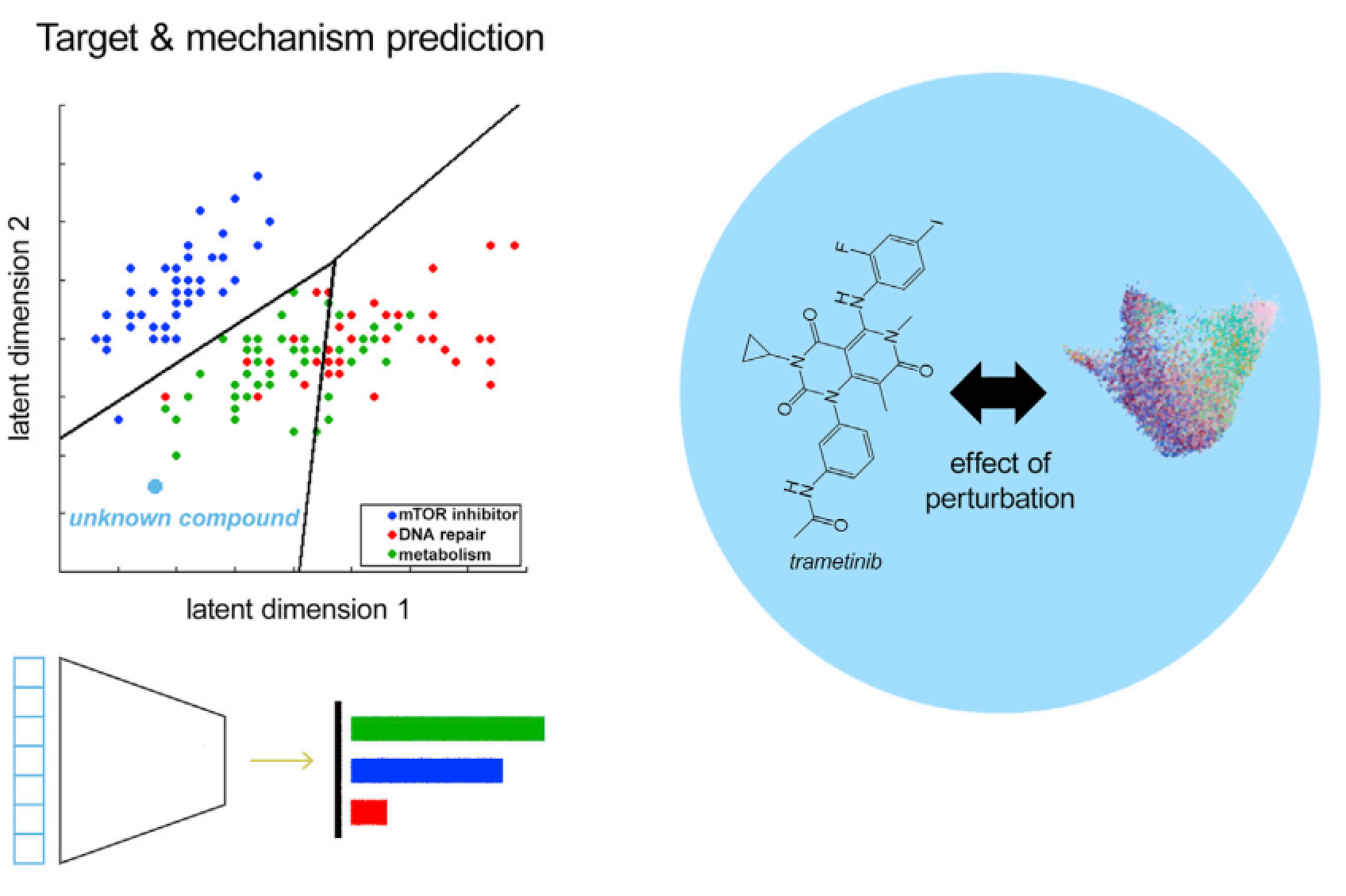
图4:通过组学数据对化合物扰动的靶点预测,红、蓝、绿分别代表模型隐空间中的具体表示
让扰动模型具有生物相关性、真正具有分子细胞生物学应用价值的主要途径就是让模型学习并预测潜在的分子机制和靶点,例如用来预测某个小分子化合物的蛋白、通路靶点。这种模型的表现可以通过对已知化合物的precision、recall等指标进行比较评估,该模型的直接应用场景就是通过对新的化合物进行评估从而发现新的药物,估计副反应,老药新用的可能性,助力药物开发。
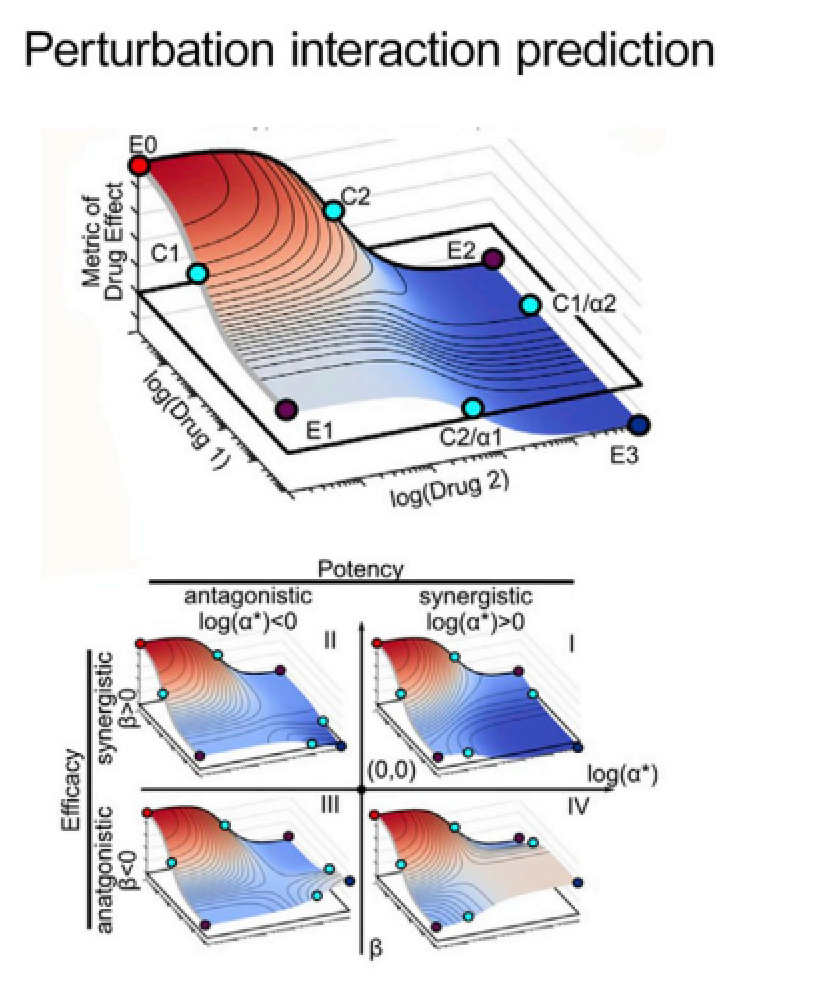
图5:预测扰动发生的联合效应,也可以理解为预测小分子化合物联合扰动所带来的效果,一些化合物对时协同性,一些是拮抗性,对于未知的化合物对,模型预测其效果
C. 扰动互作预测
扰动互作预测就是在评估成对出现的扰动效果的协同性和拮抗性(图5),化合物或者基因突变等扰动因素可以成对出现,并且可以被打上标签(协同性、拮抗姓),这种模型可以用分类或者回归模型的评价指标进行评估。该任务的应用场景同组合治疗相关,对扰动互作的了解可以更好的建立干扰互作网络,全面了解不同的扰动组合对所关心表型的影响,不同于小分子筛选,基因的knock out、knock down等实验(本质上也是一种扰动,只是发生在基因层面)可以一并在训练模型时考虑在内。
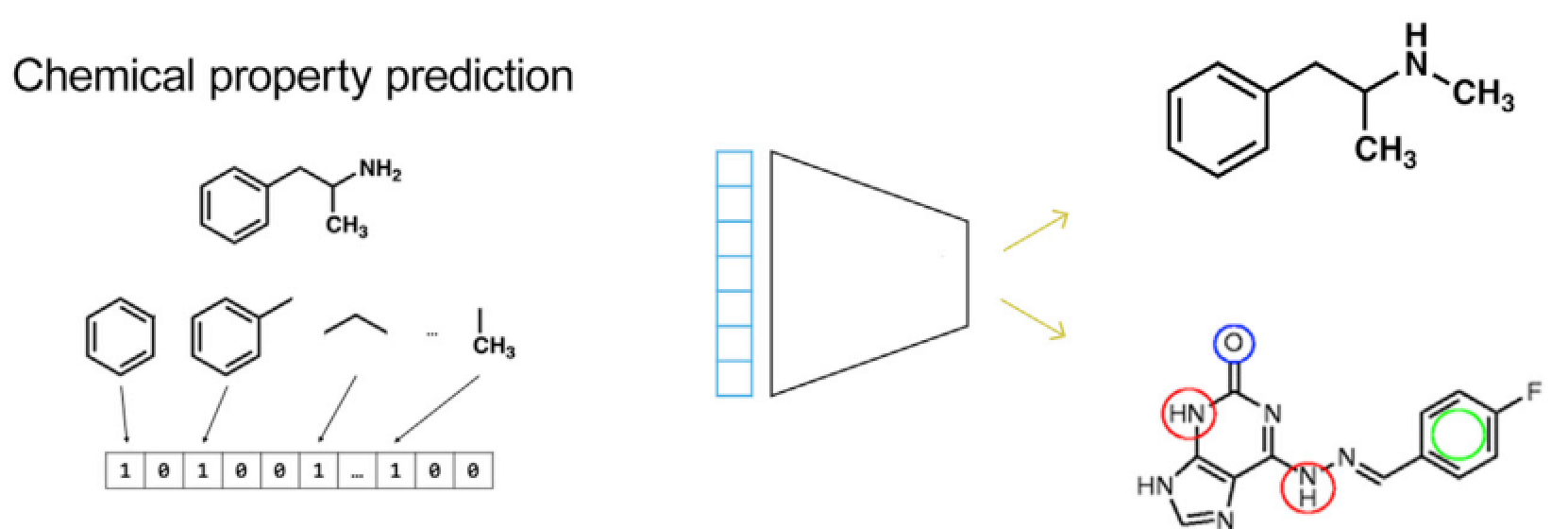
图6给定组学特征,预测扰动因素的化学特性,比如分子图谱、药效官能团或者完整的化合物
D. 化学特性预测
根据生物数据预测化合物的化学特性是连接生物学和化学的主要方式。之前的药物设计很多是根据蛋白口袋进行小分子药物的从头设计,而现在可以根据转录组学数据从头设计小分子,预测药效官能团。如果能将生物特征和化学特征联系起来,在没有进行湿实验的时候也可以对化合物的细胞毒性进行评估,对于基因层面的扰动因素,就是对k-mers、motif进行建模预测,这种模型可以在没有体外实验的时候评估该基因突变能带来的影响,甚至可以将这些数据整合起来建立网络效应。
2. 现有方法:
目前已经有很多应用于传统表观基因组、转录组和蛋白组学数据学习、分子表型筛选、临床的深度学习方法。最近的研究中,由于单细胞测序技术的大发展和单细胞数据的积累,随着CRISPR和scRNA-seq实验的相互结合,“干扰scRNA-seq”也是第一次被提出,干扰单细胞测序也成为一个独立的方向,被机器学习研究者和生物学家关注。
本文中总结了一些现有的单细胞组学模型,除了CellOracle以外,其余的模型基本都是针对干扰前后、疾病对照的实验思路进行设计训练的。因为CellOracle是利用基因调控网络(GRN)建立的,所以CellOracle的工作过程只能针对基因、单个化合物的靶点进行分析。除了如DRUG-NEM等个别模型,大部分的模型在Github上都提供有直接使用的版本和配套的安装方法和使用说明。
A. 线性回归和分类 Linear Regressor/Classifier
线性模型通过对不同表达值的线性组合同输出结果建立关系,输出可以是离散指标(如蛋白靶点)或者连续指标(如IC50)。在单细胞数据大量积累之前,这些浅层分类器或者回归模型非常常见。SCATTome就是通过线性模型来预测不同细胞状态下对药物的应答(cell-state-specific response to drug),Augur等人使用了类似的,但是非线性的随机树模型来预测这种细胞-化合物的应答。
B. 因子分解Factor decomposition
因子分解方法将组学数据分解成variation组分不同的几个部份。矩阵分解(Matrix Factorization)就是一种针对传统组学和单细胞组学常用的一种分解方法,结果容易理解并且对大数据容易扩展,通过对表达值的聚类,可以进一步进行机制方面的讨论,如信号通路、生物过程等均可进行富集分析。像上文提到的MUSIC和DRUG-NEM都是通过这种聚类,找到关键基因从而对扰动效果进行定量,或者对每一个细胞每一个蛋白特征计算概率密度,在嵌入效果模型当中使用概率矩阵来获得药物组合的效果。
C. 网络建模
网络模型是利用先验信息建立input特征之间相互关系的方法,部份干扰信息的相关数据也是缺失的,但借助这些先验知识可以对未知事件进行预测,比如已知单个药物对细胞系的影响,从而预测这些药物组合的干扰效果。CellOracle目前是唯一通过基因调控网络来利用先验知识,整合转录和转录因子调控的信息的模型。CellOracle联合了scATAC和scRNA-seq数据建立可理解的网络模型,基于该网络模型对一些未知的干扰事件进行应答预测。
3. 总结
从上述任务来看,预测扰动模型的建立就是为了捕获生物相关性,让模型去预测一些未知事件,从而减少药物开发时的费用,如果模型捕获基因干扰因素影响的特征,那么这些模型就能对病人对药物的敏感性做出解释和预测,目前通过单细胞转录组、基因组测序的方法就提供了重要的数据来源,为不同的扰动建模提供了数据支持。随着数据的大量积累,更多的研究会致力于如何建立全面的扰动全景,通过已知预测未知,透过已见寻找未见,全面深刻的理解分子生物学调控机制同扰动的关系,将是下一阶段人工智能建模在生物领域的主要方向。单细胞测序遇到人工智能,一同助力药物开发。
关于我们:
纽科生物提供专业的生物信息学数据分析和高通量测序服务。目前,公司已经和四川大学、复旦大学、上海交通大学、中山医院、华中科技大学等多所医院、高校的研究团队建立了长期良好的合作关系,提供高品质的数据分析和测序服务,帮助客户在European Journal of Heart、Circulation Research、Nature Communications等多个著名杂志期刊上发表高水平科研文章,欢迎各位老师前来咨询。