单细胞RNA-seq(single cell RNA-seq, scRNA-seq)是目前用于细胞类型、细胞状态研究的核心工具,很多实验室也同时围绕scRNA-seq技术建立了多种计算方法并且优化了建库方法,不同方法在如何鉴别分子的细胞来源和文库建立方面有所区别。scRNA-seq技术不断革新、升级的同时,随之产生的高分辨、高通量数据也助力发表了多项研究成果,以Human Cell Atlas (HCA)为代表的大型单细胞综合数据库也被建立,致力于在单细胞水平上绘制所有人类细胞转录组图谱,推动发展疾病的诊断、检测和治疗。

图1.Human Cell Atlas(https://www.humancellatlas.org/)
虽然scRNA-seq给生物研究带来了巨大的影响,但是该技术的实际应用时,仍然面临着巨大的挑战和困境。
1)单细胞悬液的制备是单细胞技术的重要环节,scRNA-seq非常依赖单细胞悬液的质量,常用的10x Genomics平台对于单细胞悬液的要求为细胞起始量大于1×10^5个,活细胞数目在85%以上,无细胞团,无碎片,因此scRNA-seq技术仅适用于新鲜组织样本,但是大量的珍贵样本都是以低温冻存的状态进行保存的,所以这类样本不适用于scRNA-seq;
2)不同的组织、样品类型的适用性不同,在单细胞悬液的制备时需要对样品解离获取单细胞状态,可是不同类型细胞的解离效率存在差异,不同组织类型的解离条件需要多次优化,像成纤维细胞、内皮细胞等多迁入细胞外基质和基底膜中,很难分离,一些敏感细胞在物理外力的处理下会发生破碎,可见不同的组织、样品类型的适用性不同;
3)部份样本类型,如心脏、肌肉、脂肪组织(含有直径比较大的心肌细胞、骨骼肌细胞、成熟脂肪细胞等),这些细胞体积大,商业化的单细胞平台对细胞大小有严格的限制,建议捕获的细胞大小不超过40um,因此30-40um的分子筛会过滤掉感兴趣的大体积细胞,有效信息会大大减少;
4)细胞本身是很脆弱的,解离过程中有可能导致细胞死亡,酶解时间,酶浓度,是否使用宽口枪头,离心力等对细胞状态均有较大影响,37度下借助物理外力的解离和酶消化过程也会给细胞状态造成很大的影响,改变细胞的生物学状态,从而在数据中引入实验造成的偏差(bias)。
酶的类型、酶解时间、酶浓度、机械损伤、离心条件、环境温度、缓冲液等均可影响单细胞悬液的制备。
单细胞核测序(single nucleus RNA-seq, snRNA-seq)对单个核进行分离测序,这种建库策略用单个细胞核代替单个细胞来表示细胞个体的基因表达水平,稳定性高且信息更加全面,细胞核可以从冰冻或者轻度固定的组织中进行提取,单核技术的这一特征有效的避免了细胞悬液制备这一过程对细胞状态的影响,很好的解决了scRNA-seq仅适用于新鲜样本的问题,本期公众号结合相关文献深度比较scRNA-seq和snRNA-seq测序技术的实际应用的优缺点。

图2. 肾脏纤维化的单核、单细胞测序技术比较https://jasn.asnjournals.org/content/jnephrol/30/1/23.full.pdf
snRNA-seq在处理罕见样本类型和发掘新的细胞状态时具有极大的技术优势。下文将从实验方法、基本信息覆盖统计、无监督聚类、等方面综合比较在肾脏组织应用结果中两种技术的具体表现。
优势1:样品前处理,snRNA-seq更为简单。
例如肾脏和其他实体样本,想要获得高质量的单细胞测序数据,技术挑战就是解离困难,如何顺利的解离细胞,保证RNA不降解,并且不引进任何因为实验造成的数据偏差,是当下首要的技术难题。本篇文章中用的scRNA-seq和snRNA-seq高通量技术的样本前处理的大致流程如下。
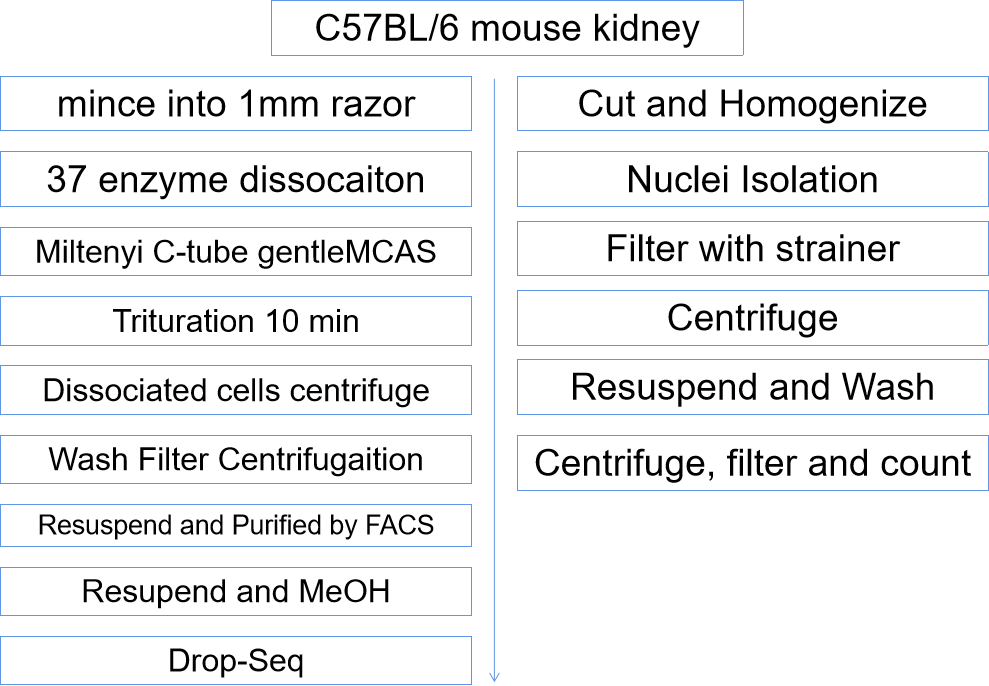
图3. 单细胞和单核样品处理的实验流程简图
实验方法1:单细胞解离+甲醇处理
C57BL/6小鼠肾脏切割为1mm大小组织块,250U/ml Liberase和40U/ml DNaseI解离缓冲液在37摄氏度酶解,之后进行10min研磨消化,使用10%FBS终止消化,分离得到的细胞500x 5min离心,洗两遍,过35um滤网,然后离心500x 5分钟,PBS中细胞重悬,添加1%BSA,FACS纯化,然后使用FACS进行前向角散射(forward scatter)和侧向角散射(side scatter),每个肾脏收获约2百万个细胞,用甲醇处理,200ul冷PBS重悬,预冷100%甲醇缓慢滴入细胞悬浊液中进行固定,7天存储后PBS从-80移动到冰上,PBS洗两遍过40um过滤网,技术,稀释预处理用于DropSeq。
实验方法2:单核分离
使用添加有蛋白酶抑制剂和RNase酶抑制剂和Nuclei EZ裂解缓冲液(NUC-101;Sigma-Aldrich)捕获单个细胞核,样品分割为<2mm大小,使用Dounce Homogenizer在裂解缓冲液中均一化,过40um过滤网,离心,重悬洗涤,过20um过滤网后计数。
优势2:信息覆盖snRNA-seq更全面。
scRNA-seq使用scDropSeq方法实现,而snRNA-seq通过snDropSeq\DroNc-seq\sn10X获取。
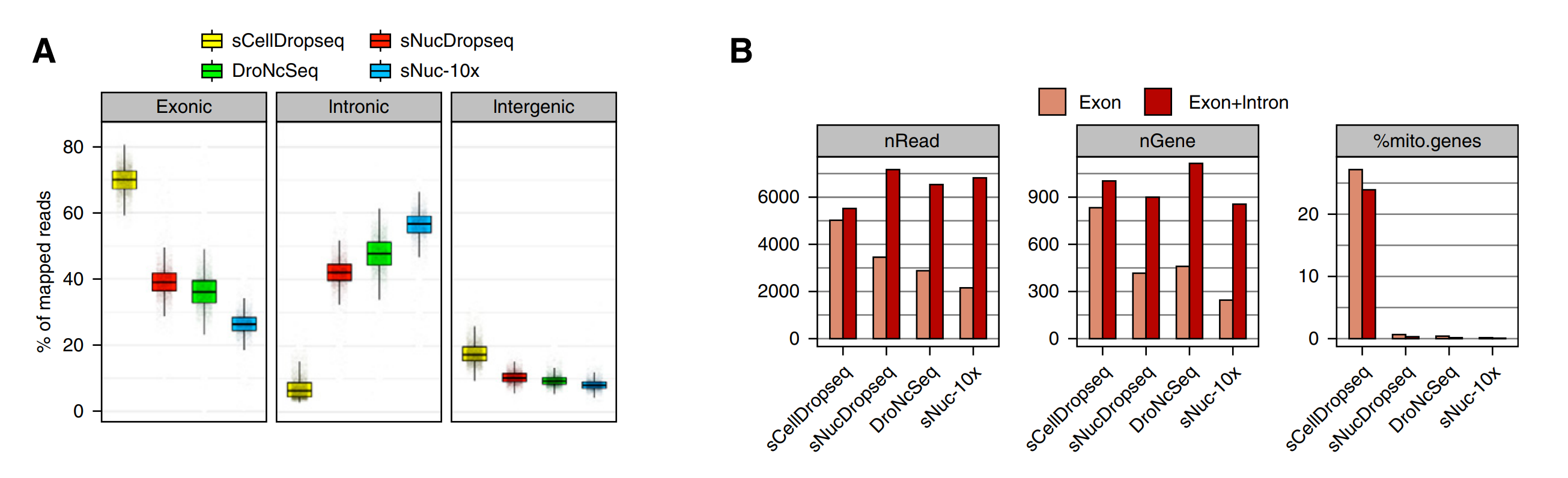
图4.snRNA-seq核scRNA-seq的reads匹配比较
scRNA-seq和snRNA-seq产生的数据各自有什么特点?
1)scRNA-seq产生的数据大部分比对到外显子区域,而snRNA-seq多数匹配到内含子和基因间区,虽然单细胞数据更多的覆盖到外显子区域;
2)因为线粒体是独立于细胞核独立存在的细胞器,所以只是在scDropSeq数据中检测到线粒体来源的RNA信息,占到了整个细胞的24%,这一部分信息时scRNA-seq独有的;
3)虽然细胞核的mRNA含量远远低于整个细胞,但是单核测序平均每个细胞的基因表达数量和整个细胞的基因表达数目相当,把这部分信息除去以后snRNA-seq的基因检出率反而要高于scDropSeq数据,尤其在基因表达检测方面相当甚至由于单细胞测序。
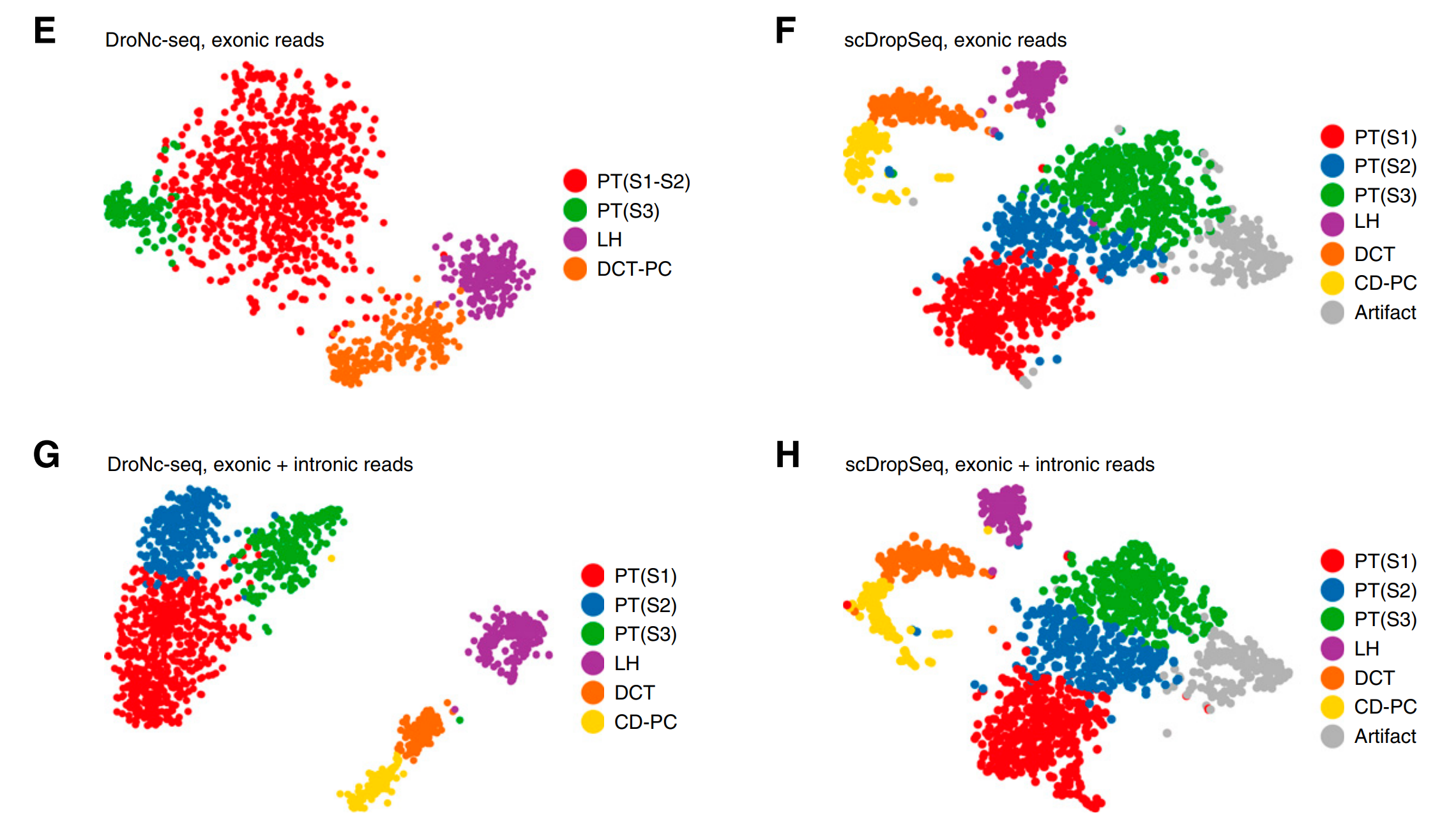
图5.无监督聚类结果比较
优势3:snRNA-seq 聚类效果更佳。
1)在无监督聚类的结果上比较snRNA-seq和scRNA-seq的表现。如果仅分析外显子匹配得到的reads信息,单核测序结果可以找到4个cluster,单细胞测序结果可以找到7个cluster;再加入内含子信息的时候,单核测序可以找到6种细胞类型,单细胞测序的细胞亚群数目没有改变,可见当充分应用内含子匹配到的reads信息时,snRNA-seq在无监督聚类的分类数目同scRNA-seq相当;
2)scRNA-seq聚类分析得到的7个细胞压群中,有一个细胞亚群(Artifact,对应图5H的灰色亚群)高表达压力应答基因,这些基因的高表达实际上是由于细胞解离时因为实验操作引入的偏差,snRNA-seq消除了这一部分错误的信息,其次在组织解离时由于细胞破裂从而导致释放到环境中的mRNA能够污染单细胞测序数据,在其他发表的论文中,相关数据分析也发现在几乎所有的cluster中Slc34a1基因都是高表达的,虽然在单核测序数据中也发现了类似的情况,但是相比于scRNA-seq数据,单核测序的污染程度已经被大大缓解;
3)引入内含子reads的时候可以提高snRNA-seq聚类结果中不同类内部的cohesion(拉近同一个cluster内样品的基因表达平均值)并且增加不同亚群之间的间隔(separation),但是对于单细胞测序分析来讲,对聚类的结果没有显著改变;
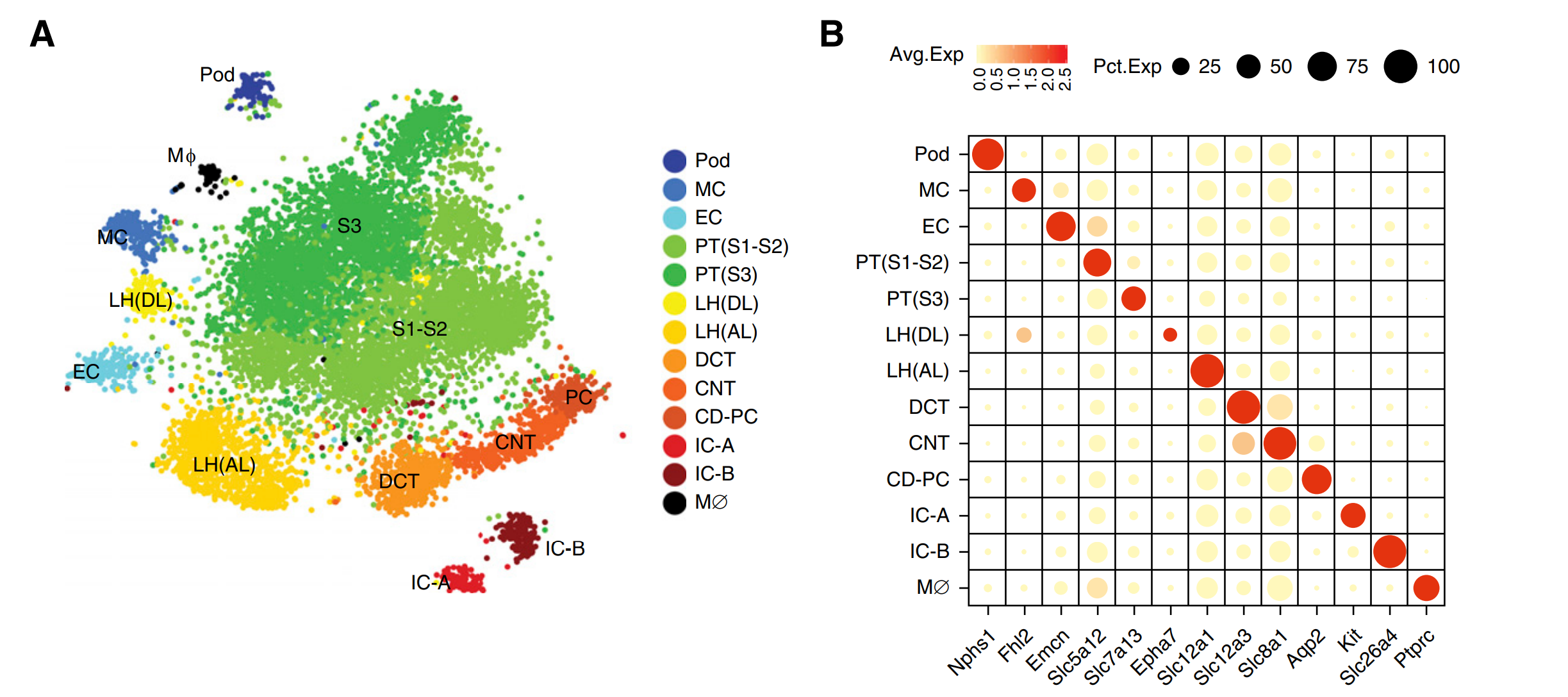
图6 整合分析和Marker基因的相关性分析
单细胞与单核测序数据进行整合分析,去批次效应,t-SNE发现了包括足细胞、内皮细胞、肾小球膜细胞、肾小管细胞和巨噬细胞在内的共13个cluster,和其他最近发表的肾脏单细胞测序数据比较分析,对细胞亚群的注释结果进行确认。
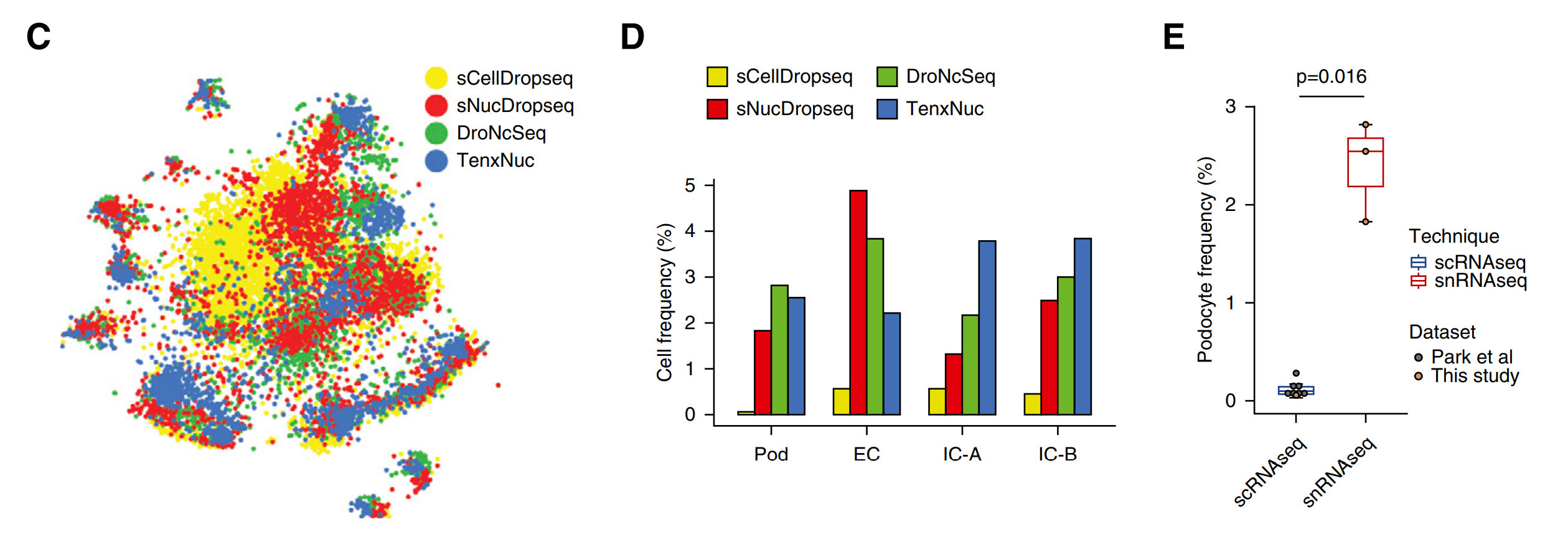
图7 整合分析和Marker基因的相关性分析
优势4:snRNA-seq 在整合分析中占主导作用,部分细胞的检出灵敏度更高,结果可靠
tSNE结果能分析每个平台数据对每个cluster的贡献度,图7C当中显示,红色、绿色、蓝色所代表的单核测序数据的贡献程度均高于黄色的单细胞数据,这三种测序平台在检测足细胞、内皮细胞、闰细胞(intercalated cell)方面有更高的灵敏度,从图7D中看到,EC、IC-A、IC-B三种细胞的红、绿、蓝色代表的单核测序数据对应细胞的检测频率要明显高于黄色的单细胞测序平台,图4E的结果显示,snRNA-seq结果中的足细胞检出率是scRNA-seq对应检出率的20倍(2.4% vs. 0.12%,P=0.02)。
在基因差异表达方面, 9588(71.4%)个表达的基因在scRNA-seq和snRNA-seq中的表达情况类似,有676个基因(5%)在单细胞数据中高表达(fc 1.5;P<0.05),863(6.4%)个基因在单核数据中高表达。scRNA-seq数据中高表达基因功能分析显示,这些基因主要包括线粒体、核糖体、热休克信号通路相关,值得一提是的在snRNA-seq中高表达的基因是和一些cell identity、转录因子相关的基因,和最近发表的有关脑组织中的富集分析结论一致。同样在单核数据中能够检测到更多的长链非编码分子。
这些表达差异是否会影响细胞聚类结果?从snDropSeq和DroNc-seq我们提取了足细胞、内皮细胞、系膜细胞共650个单核,通过随机森林算法在肾小球单细胞图谱(glomerular single-cell atlas)中选取了最相似的650个最匹配的单细胞整合分析,所有细胞可以聚类成三种独特的细胞类型,通过MetaNeighbor方法来预测scDropSeq得到的细胞亚群。尽管一些基因的表达数据有所差别,但是snRNA-seq基本能够以较高置信度的完成细胞分类。
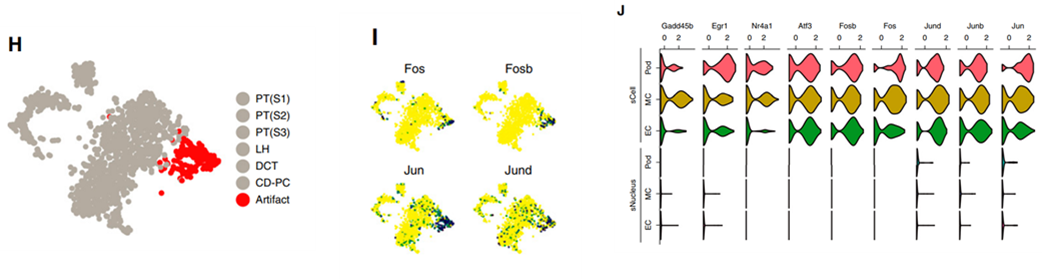
图8 压力应激基因在snRNA与sCell中的相关表达
优势4:snRNA-seq 高信噪比
37摄氏度下的组织解离过程确实诱导一些压力应激基因的表达,在scRNA-seq数据的当中,有一个细胞亚群就是基于压力应答基因进行分类获得的,整个抽核过程是在冰上进行,这样可以成功的避免转录组数据的改变,我们在对glomerular atlas数据重分析时候发现,几乎所有细胞也都存在丰富的压力应答基因的广泛高表达(图8J),但是在snRNA-seq数据中无明显表达。
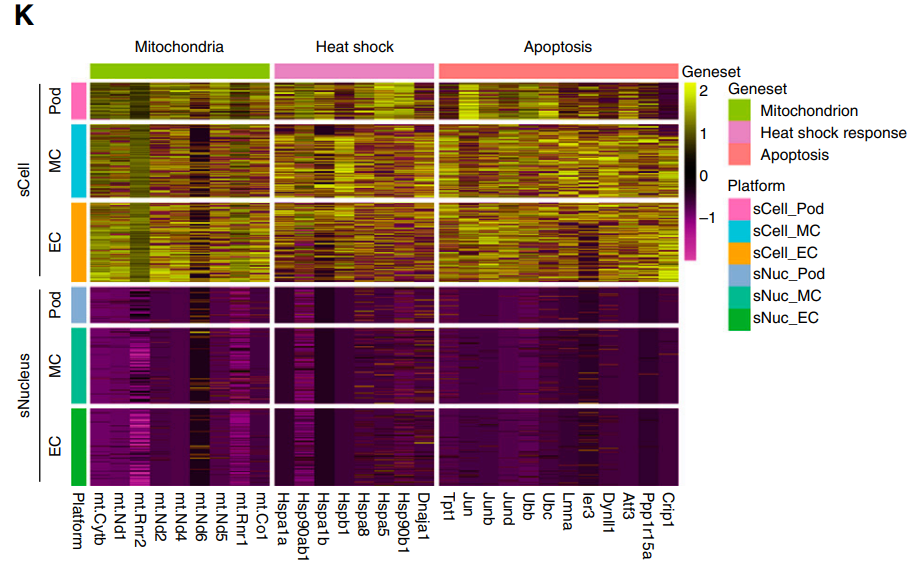
图9 肾小球细胞snRNA-seq和scRNA-seq在线粒体、热休克、凋亡相关基因的表达差异
通过对glomercular 细胞类型的差异基因分析可以发现,线粒体基因、热休克基因、细胞凋亡基因在scRNA-seq平台中都会呈现高表达,而在snRNA-seq数据中,这类基因的表达丰度相对较低。
优势4:snRNA-seq分析罕见细胞类型和细胞亚群的发现
接下来使用纤维化炎症肾脏组织对单核数据进行验证,一共发现在sn10X平台上产生的数据检测平均每个单核有763个基因1206个分子标记,无监督聚类的结果发现17个独特的细胞亚群,在这些结果中发现了两类新的近端小管(proximal tubule)细胞亚群,其中一类高表达增值相关的基因(proliferative gene signature),因此将这类基因注释为proliferative 近端小管细胞(Prolif.PT),与此同时这类细胞还表达损伤相关的marker基因,比如Havcr1和Vcam1。另一类新的近端小管(proximal tubule)高表达细胞运动相关基因,有趣的是这类细胞亚群也表达部份损伤相关基因,如Vcam1,但不表达Havcr1,同时还高表达许多分泌促炎细胞因子,包括巨噬细胞趋化Ccl2、巨噬细胞因子 Il34和中性粒细胞趋化因子Cxcl1和Cxcl2,定义为dedifferentiated近端小关细胞(Dediff.PT)。
差异基因分析显示proliferating cluster主要富集一些细胞周期相关的条目,dedifferentiated cluster主要是细胞运动调控相关的条目,例如rhoGEF Dock10主要在dedifferentiated cluster中富集表达,这个基因调控通过Cdc42信号通路进行细胞形态塑造(morphogenesis)。
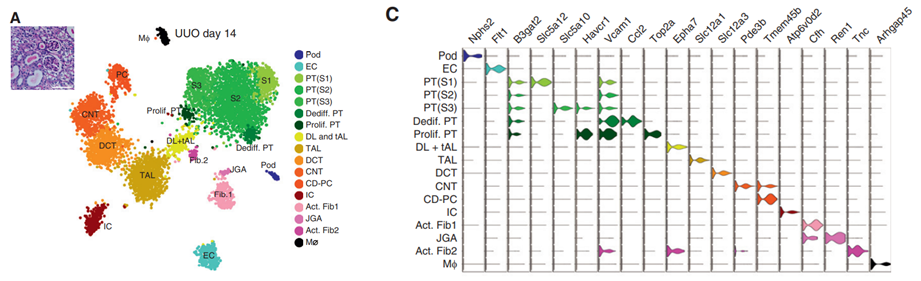
图10 t-SNE肾脏snRNA-seq分析与Marker基因的表达
1)基于Ren1和内皮素受体A表达找到了一些罕见的球旁复合体(juxtaglomerular apparatus) 细胞,而这类细胞也表达Hopx基因;
2)新发现了两群独特的活化的成纤维细胞,其中一群表达2型甘露糖受体,这种分子和结合胶原减轻肾纤维化,另外一类活化的成纤维表达tenascine C,在最近的研究认为这类分子是细胞外基质磷酸糖蛋白,能够促进肾纤维化,这两类细胞都能表达平滑肌肌动蛋白。
3)snRNA-seq数据的细胞通讯分析能发现一些新的细胞间交流的信号通路,分析显示像已知的Bmp6、Pdgfd、Spp1和新的Sema3c、Sema6a、Gpc和Slit3信号通路,图11G就总结了纤维化肾脏的肾小管间质互作网络。
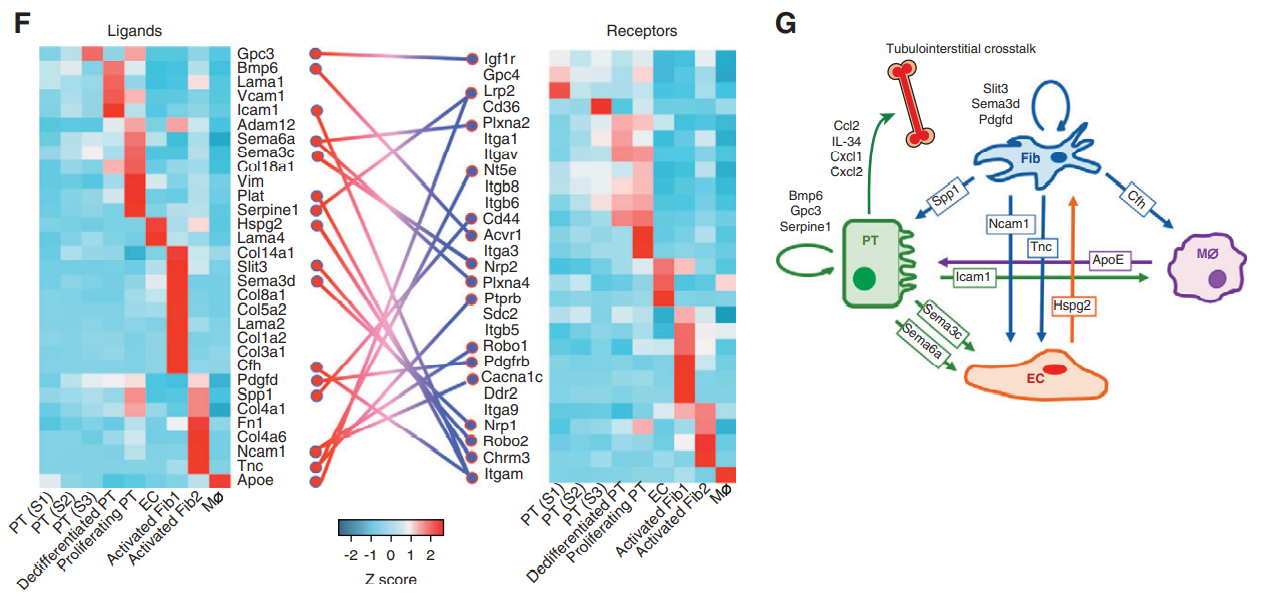
图11 受体配体相互集合与细胞间信号传导通路
总结:snRNA-seq实验过程中避免因为解离细胞带来的实验偏差,但是在基因表达检测方面可以提供与单细胞技术同等的效能,这一点非常重要,相比于单细胞测序,单核实验过程减少了所需的细胞数量同时增加了有效信息量。使用snRNA-seq技术在肾纤维化组织上确实发现了新的细胞状态和一些罕见的细胞类型,并且成功构建了更加完整的细胞通讯网络,细粒度的分析并理解了肾脏纤维化的生物学过程。总体来看,snRNA-seq的优势在于样品处理过程更加简单、测序信息覆盖更加全面、细胞亚群聚类效果更好、信噪比、灵敏度高且结果可靠 ,针对罕见样本和发现新的细胞状态和细胞亚型具有明显的技术优势。
纽科生物在单个细胞核实验积累了丰富的经验,采用高信噪比试剂,高质量的完成对单个细胞核的提取与测序,实验流程进行反复优化,欢迎各位老师前来咨询纽科生物冷冻组织单核ATAC测序与冷冻组织单核转录组测序相关产品。
关于我们:
纽科生物提供专业的生物信息学数据分析和高通量测序服务。目前,公司已经和四川大学、复旦大学、上海交通大学、中山医院、华中科技大学等多所医院、高校的研究团队建立了长期良好的合作关系,提供高品质的数据分析和测序服务,帮助客户在European Journal of Heart、Circulation Research、Nature Communications等多个著名杂志期刊上发表高水平科研文章,欢迎各位老师前来咨询。